Unwavering Support for Healthcare Providers
2026/02/25
2026/03/02
Author: Dr. Evelyn Reed, MD
Lead Medical Content Reviewer & Clinical Advisor at VistaMed Technologies
Dr. Reed specializes in evaluating the clinical evidence behind medical devices, ensuring that every claim is supported by robust data that stands up to the highest regulatory scrutiny.
I was recently reviewing the technical file for an older pulse oximeter that had been certified under the previous EU directive. The clinical evaluation report was barely ten pages long, based on a handful of small, dated studies. By contrast, the Clinical Evaluation Report (CER) our team prepared for the VistaMed FPO-50 under the new regulation is over 100 pages. It's a living document, cross-referenced with risk management files, usability engineering reports, and a proactive plan for post-market data collection.
The level of scrutiny is night and day. And for a medical device distributor, understanding this difference is no longer an academic exercise. It is a fundamental question of commercial survival in the European market.
For years, distributors operated on a simple premise. But the ground has shifted beneath our feet, creating a dangerous myth.
The Myth: A device with a CE mark is legal to sell in the EU, and my main job is to find the best unit price.
The Reality: The old Medical Device Directive (MDD) has been fully replaced by the far stricter Medical Device Regulation (MDR 2017/745). The transition period is over. Devices holding legacy MDD certificates are losing their legal status for being placed on the market. If you are a distributor holding stock of oximeters certified under the old MDD, you are sitting on a ticking clock. Your inventory could become unsellable overnight.
So, what does it actually take to get an MDR CE mark for a device like the FPO-50 Fingertip Pulse Oximeter? The new regulation moves away from a one-time approval to a lifecycle approach. As the person who reviews these documents, the difference is stark. A CER under MDR isn't a snapshot; it's a motion picture, constantly updated with real-world performance data from our Post-Market Surveillance (PMS) plan.
For a distributor, this means your manufacturing partner is legally mandated by EU law to be proactive about quality, not reactive. They must actively collect and analyze data on their device's performance after it's sold. This is your assurance that you are not the last line of defense for product quality—the manufacturer is.
The demanding MDR process results in a fundamentally better, more reliable product. The differences aren't always visible on the outside, but they are critical to the device's performance and your ability to sell it to discerning clinical customers.
|
Feature |
"Old" MDD-Era Oximeter |
VistaMed FPO-50 (MDR Certified) |
Why This Matters to a Distributor |
|
Certification Basis |
One-time technical file review under MDD |
Lifecycle evidence under MDR 2017/745 |
Guarantees ongoing legal market access in the EU. Protects your inventory from becoming obsolete. |
|
Clinical Data Depth |
Often relies on equivalence to older devices. |
Requires fresh, robust clinical data for the specific device. |
Provides your sales team with powerful, evidence-based claims to win over hospital clients. |
|
Displayed Metrics |
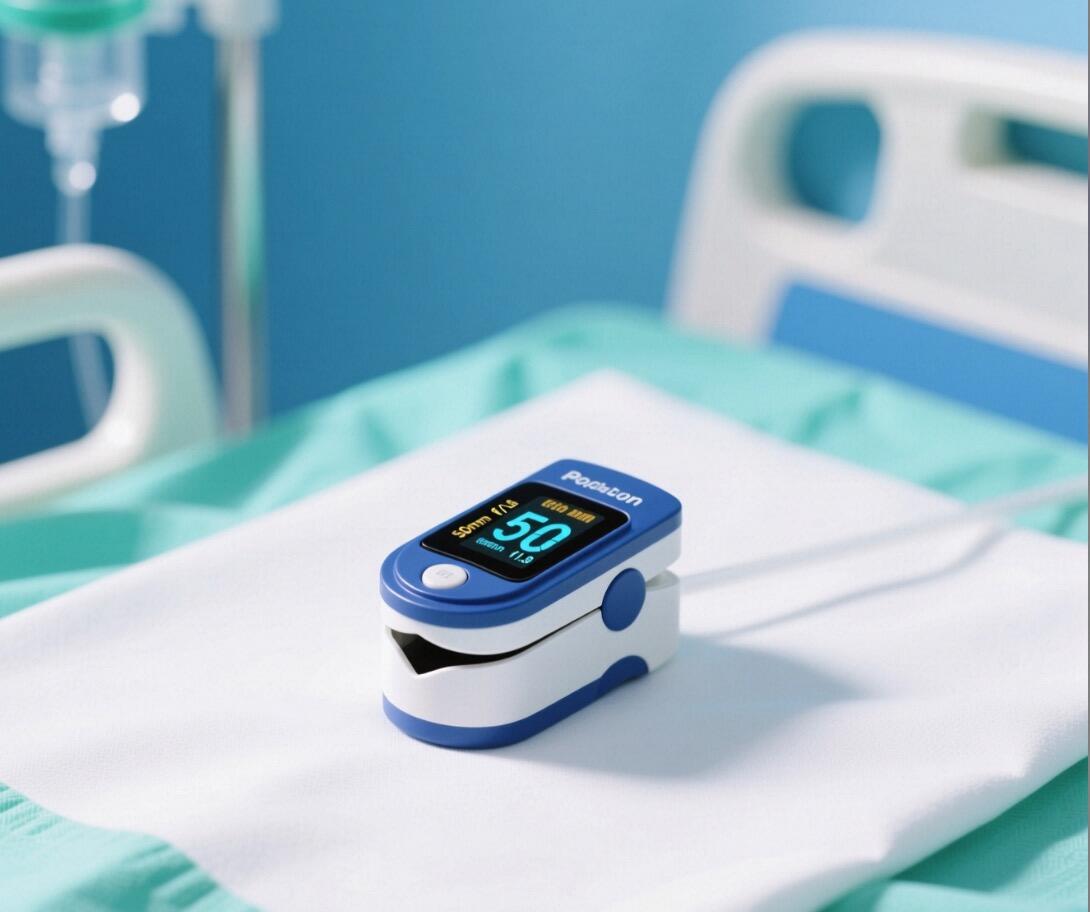
SpO₂ and Pulse Rate only. |
SpO₂, Pulse Rate, and Perfusion Index (PI). |
The PI is a key differentiator, indicating the reliability of the reading and demonstrating superior technology. |
|
Post-Market Obligation |
Reactive (complaint handling). |
Proactive (scheduled PMS reports, trend analysis). |
Reduces your risk. The manufacturer is actively monitoring for issues before they become your problem. |
From a clinical perspective, a feature like the Perfusion Index (PI) is critical. It’s a numerical value that indicates the strength of the blood flow in the finger where the reading is being taken. A low PI means the signal is weak, and the resulting SpO₂ value may be unreliable. Many older, cheaper devices omit this metric entirely, creating a false sense of confidence in potentially bad data. For your hospital clients, that's a patient safety issue. Being able to explain this to a clinical buyer instantly elevates you from a box-shifter to a knowledgeable consultant.
"An MDR-certified partner isn't just selling you a compliant device; they are offering you insulation from regulatory risk. They have done the incredibly expensive and time-consuming work—work that costs millions and takes years—so you can focus on sales, confident that the product you're placing won't be pulled from the market." – Dr. Evelyn Reed, MD
This commitment to data integrity isn't just a European quirk; it's a global trend. A manufacturer who meets the high bar of EU MDR is better positioned to meet rising standards everywhere, future-proofing your business.
For instance, the US FDA is now intensely focused on the issue of pulse oximeter accuracy across different skin pigmentations, a concern raised in a public advisory committee meeting. Addressing this requires exactly the kind of robust, diverse clinical data sets that are mandated under the EU MDR. By partnering with an MDR-compliant manufacturer, you are aligning with a supplier who is already prepared for the next wave of regulatory scrutiny in the U.S. and other major markets.
It's this level of validated performance that leads top-tier research institutions to select devices for their critical work. Our commitment to clinical validation is demonstrated through collaborations, such as a remote monitoring study with the Cardiovascular Research Institute at Stanford University, which was published in the Journal of Telemedicine and Telecare. Projects like this demand the highest possible confidence in data accuracy—the very same confidence you should demand from your supplier.
About the Author
Dr. Evelyn Reed, MD serves as Lead Medical Content Reviewer & Clinical Advisor at VistaMed Technologies. With over a decade of experience in medical communications, she specializes in translating complex clinical data and technical information into clear, accurate, and actionable insights for healthcare professionals. At VistaMed, Dr. Reed is responsible for the final medical review of our clinical evidence pages, product guides, and educational materials, ensuring every claim is supported by evidence and presented with the utmost clarity and integrity. Her analysis in this article stems from her direct, in-depth reviews of regulatory technical files and clinical evaluation reports under both the old MDD and new MDR frameworks.
Clinically & Regulatory Reviewed By: Jian Wang (王健), RAC, Vice President, Quality & Regulatory Affairs
The information provided is for informational purposes and intended for a B2B audience of healthcare professionals and procurement decision-makers. It is not a substitute for professional medical or financial advice. TCO and ROI results may vary based on facility size, usage patterns, and local market conditions. All certifications and regulatory clearances referenced are accurate as of the date of publication. Please contact VistaMed Technologies for the most current documentation.

2026/02/25